

¿Cuáles son los síntomas más frecuentes?
Las alteraciones cognitivas en la EH pueden ocurrir varios años antes del diagnóstico (la aparición de los síntomas motores) y se deterioran de manera progresiva a medida que avanza la enfermedad.
Las manifestaciones conductuales en la EH son particularmente diversas y también pueden ocurrir muchos años antes de que se realice un diagnóstico clínico de la patología 15-18. Los síntomas motores en la EH son inicialmente sutiles y progresan en una trayectoria no lineal a lo largo de la enfermedad 8-19.
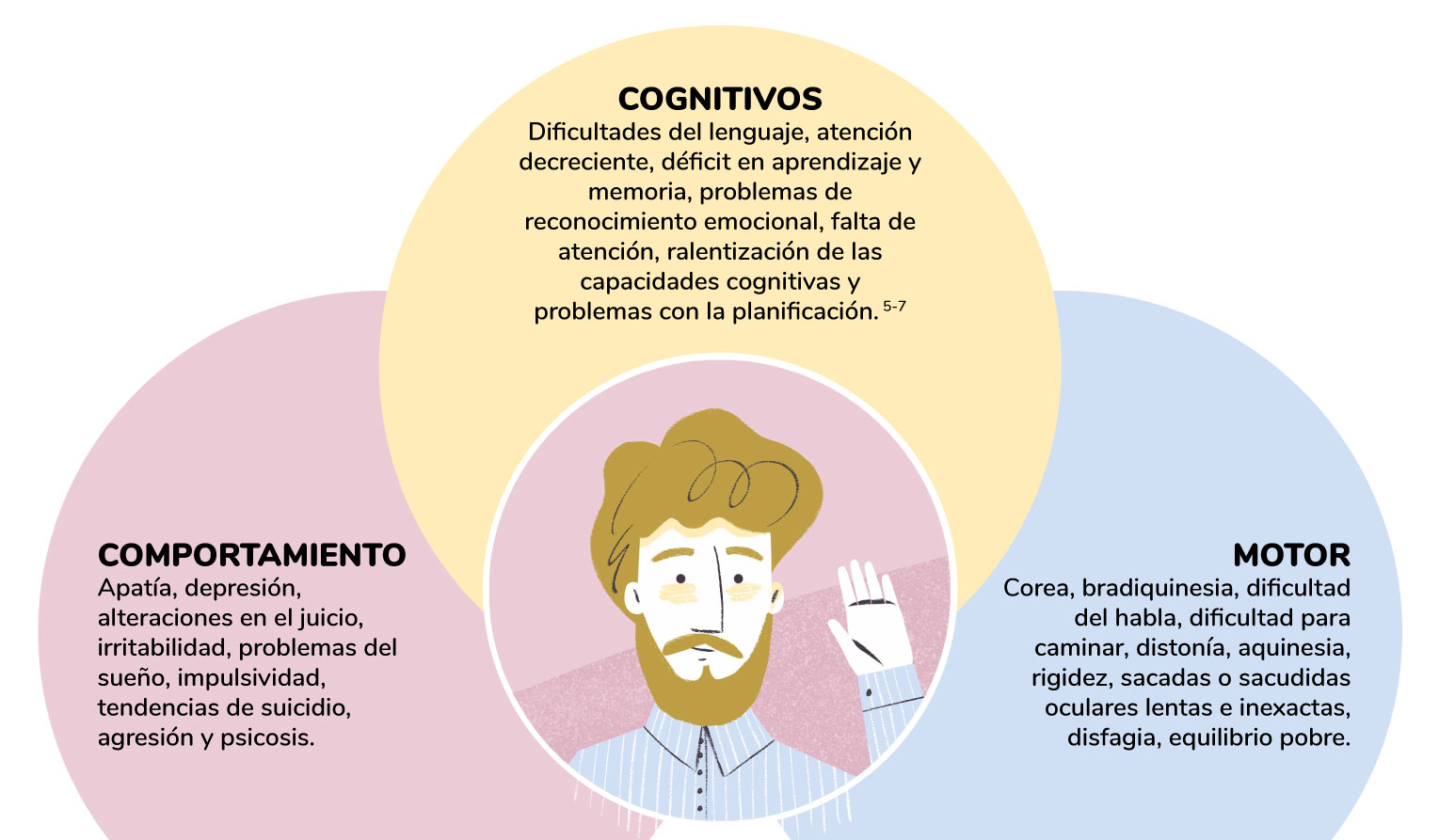
* Esta no es una lista completa de los síntomas de la EH. No todos los pacientes presentan todos los síntomas. Los signos y síntomas difieren para cada individuo con la EH.
Dado que las personas experimentan estos síntomas de una manera única e individual, la EH a menudo puede ser difícil de diagnosticar. Esto significa que también puede ser un desafío para la atención del paciente, por lo que un enfoque multidisciplinario es fundamental y puede ayudar a mejorar el manejo de la enfermedad.
Etapas de la patología
El diagnóstico clínico de la EH se define típicamente por la aparición de síntomas motores inequívocos, generalmente entre los 30 y 50 años de edad 7-8-21-22. La terminología de etapas de la EH sigue evolucionando; sin embargo, la enfermedad generalmente progresa a través de las siguientes etapas 5-7-22:
1. EH Premanifiesta:
Personas que portan la mutación del gen causante de la EH pero que aún no han presentado síntomas motores 6-8.
1.a. Presintomática: Personas con EH pero que aún no han desarrollado ningún síntoma.
1.b. Prodrómica: Los individuos experimentan cambios sutiles en la cognición, el estado de ánimo y el comportamiento. Estos síntomas aparecen años antes del diagnóstico o la aparición de signos motores inequívocos 6-8-21-23. Los cambios cerebrales, incluida la atrofia estriatal, pueden evidenciarse en esta etapa de la enfermedad 6.
2. EH manifiesta:
Personas que portan la mutación del gen causante de la EH, con presencia de síntomas motores inequívocos 8.







