
¿Qué es y qué origina la Atrofia Muscular Espinal?
La Atrofia Muscular Espinal (AME) es una enfermedad neuromuscular de carácter hereditario, que se manifiesta por una pérdida progresiva de la fuerza muscular1, y puede afectar actividades esenciales como hablar, respirar, caminar o tragar.
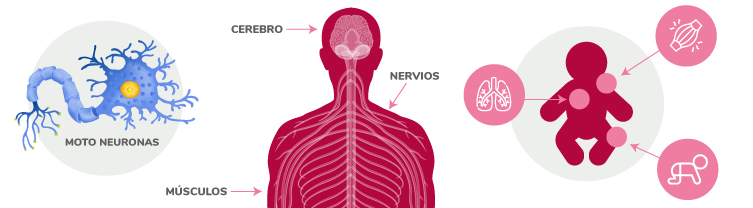
El movimiento muscular voluntario se produce gracias a la transmisión y recepción de señales que envían las neuronas motoras. Cuando hay interrupciones en estos impulsos nerviosos, se produce debilidad muscular y atrofia.
La enfermedad se debe a la mutación del gen SMN1, responsable de la supervivencia de las motoneuronas.
En AME existen defectos en el gen SMN1, el encargado de la fabricación de la proteína SMN, y al presentarse niveles insuficientes de esa proteína, las neuronas motoras dejan de funcionar con normalidad.
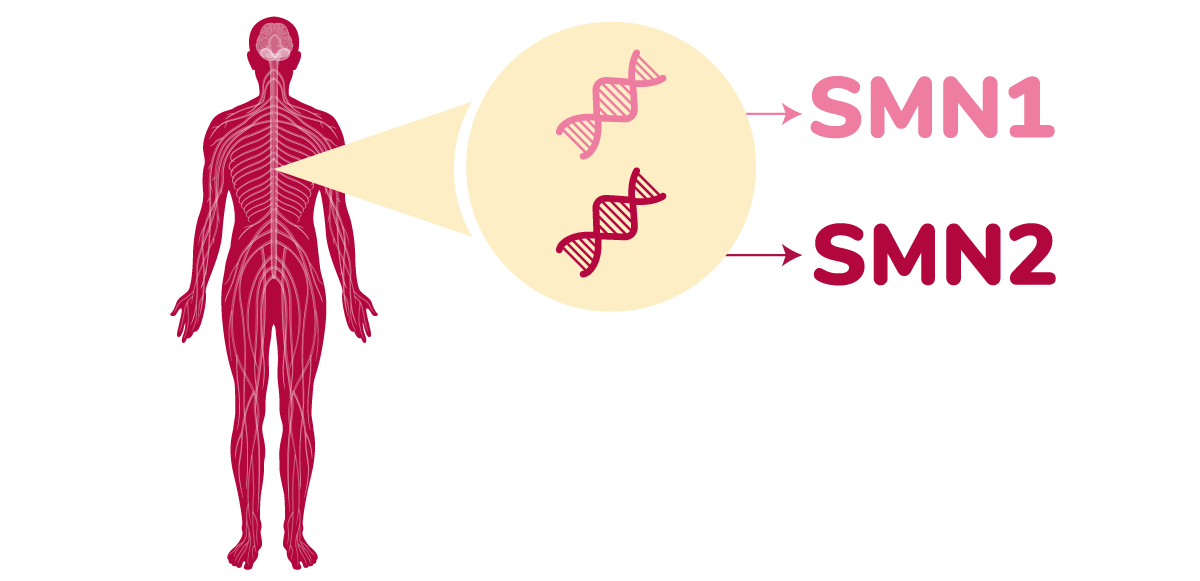
- El cuerpo utiliza dos genes estructuralmente parecidos para elaborar la proteína SMN necesaria para la función de la motoneurona.
- El gen SMN1 produce prácticamente toda la proteína SMN necesaria mientras que el gen SMN2 sólo produce una pequeña parte (10% aproximadamente).
- Las motoneuronas transmiten las señales del cerebro a los músculos y controlan la fuerza muscular y el movimiento. Su alteración por esta enfermedad, impide transmitir señales cerebrales, produce debilidad y atrofia de los músculos.
- Las personas que tienen esta enfermedad, pueden ver afectada su capacidad para gatear, sentarse, caminar, controlar los movimientos de la cabeza y hablar. En los casos graves, la afectación de los músculos puede limitar la respiración y la deglución.
Tipos de AME y edad de aparición
Aunque la Atrofia Muscular Espinal es diagnosticada principalmente durante la infancia, puede afectar a personas de cualquier edad, incluso adultas.
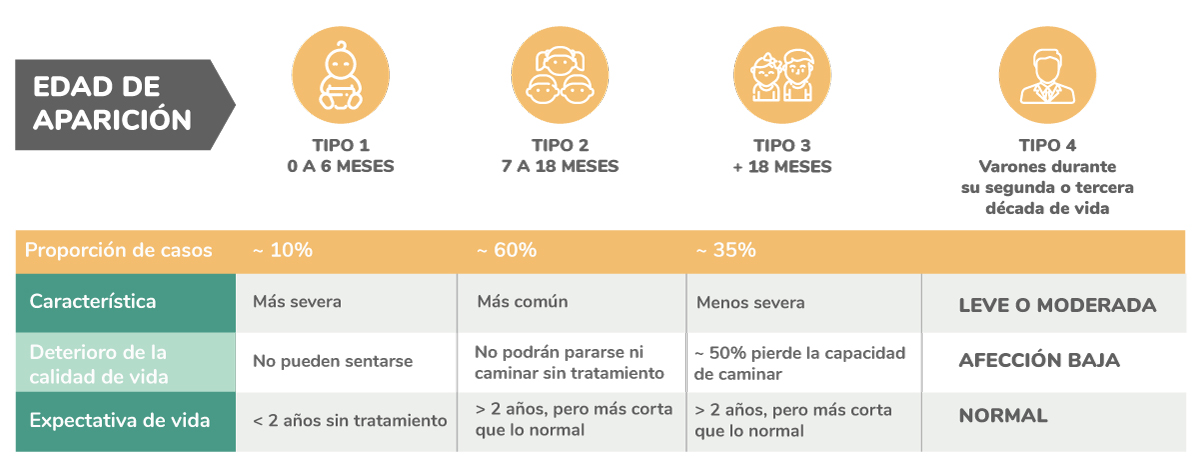
- AME tipo I (también denominada enfermedad de Werdnig-Hoffmann o AME de inicio infantil): se evidencia en los 6 meses de edad. Los niños afectados no pueden sentarse ni pararse. Se estima que, sin tratamiento, la esperanza de vida es muy limitada. Algunos síntomas son: disminución del tono muscular, espasmos, temblores, dificultad para respirar, entre otros.
- AME tipo II (también se la conoce como AME intermedia): los síntomas generalmente comienzan entre los 6 y los 18 meses de edad. Los niños pueden llegar a sentarse sin apoyo, pero no logran pararse o caminar sin ayuda. La expectativa de vida es reducida, pero con tratamiento pueden llegar a la adolescencia o a ser jóvenes adultos.
- AME tipo III (enfermedad de Kugelberg-Welander): aparecen entre los 2 y los 17 años de edad. Los niños pueden llegar a caminar pero, si no llevan adelante un tratamiento, muchos perderán su capacidad de hacerlo. Los síntomas incluyen marcha anormal, dificultad para correr y/o inconvenientes para subir escalones, entre otros.
- AME tipo IV (Síndrome de Kennedy o AME Bulbo Espinal): aparece en varones durante su segunda o tercera década de vida. Se presenta de forma leve o moderada. En este caso, los músculos respiratorios y de deglución no suelen estar afectados.2
¿Cómo se transmite?
La enfermedad de AME se pueden heredar cuando ambos padres son portadores del gen SMN1 defectuoso, lo que se conoce como transmisión recesiva autosómica3. De todos modos, la probabilidad de que cada hijo exprese la enfermedad es de un 25%.
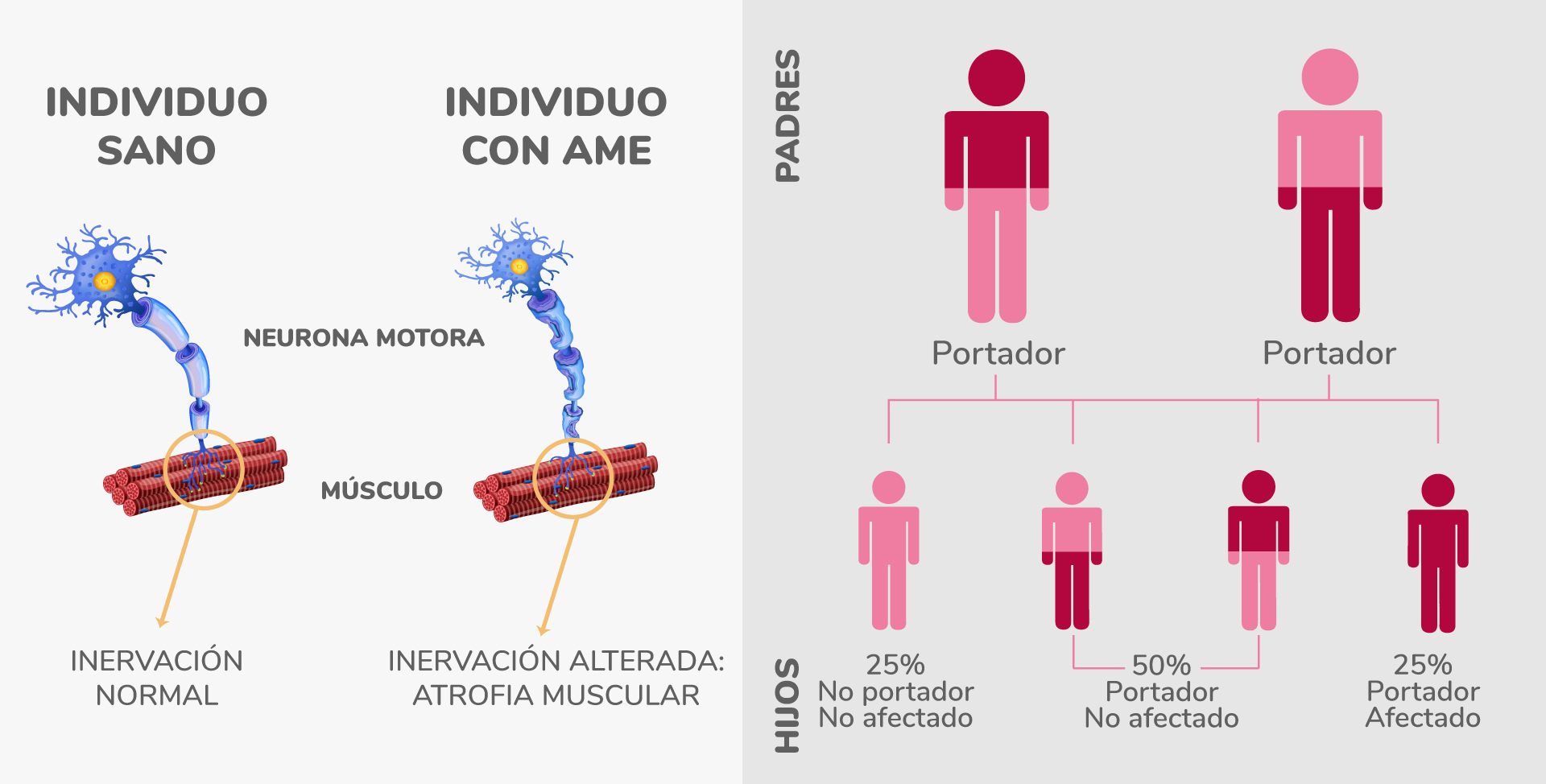
Quienes portan solo un gen mutado son portadores de la enfermedad sin tener ningún síntoma. Las enfermedades autosómicas recesivas pueden afectar a más de una persona en la misma generación (hermanos o primos).3
Incidencia y búsqueda de acciones terapéuticas
Esta enfermedad tiene una incidencia de aproximadamente uno de cada 6.000 recién nacidos vivos en el mundo4.
La incidencia de esta enfermedad es relativamente baja, pero entre las enfermedades genéticas graves con herencia autosómica recesiva es la segunda en frecuencia después de la fibrosis quística y es la causa más frecuente de muerte genéticamente determinada5.
¿Cómo se diagnostica la Atrofia Muscular Espinal (AME)?
El diagnóstico de sospecha de AME, sobre todo en las formas infantiles, es generalmente clínico (historia de dificultades motoras, examen físico en el que se evidencia la alteración de las neuronas motoras), pero es el análisis genético el que confirma el diagnóstico.
Se encuentra disponible un análisis de sangre para buscar deleción* o mutaciones del gen SMN1.
Esta prueba identifica al menos el 95 por ciento de los tipos de AME I, II y III y también puede revelar si una persona es portadora de un gen defectuoso que podría transmitirse a los niños.
Si no se encuentra que el gen SMN1 sea anormal o el historial y el examen del individuo no son típicos de la AME, otras pruebas de diagnóstico pueden incluir electromiografía que registra la actividad eléctrica de los músculos durante la contracción y en reposo, estudios de velocidad de conducción nerviosa que miden la capacidad del nervio para enviar una señal eléctrica, y otros análisis de sangre.
*Deleción: tipo de mutación genética en la cual se pierde material genético, desde un solo par de nucleótidos de ADN hasta todo un fragmento de cromosoma. Fuente: Genome.gov.ar
Algoritmo Diagnóstico
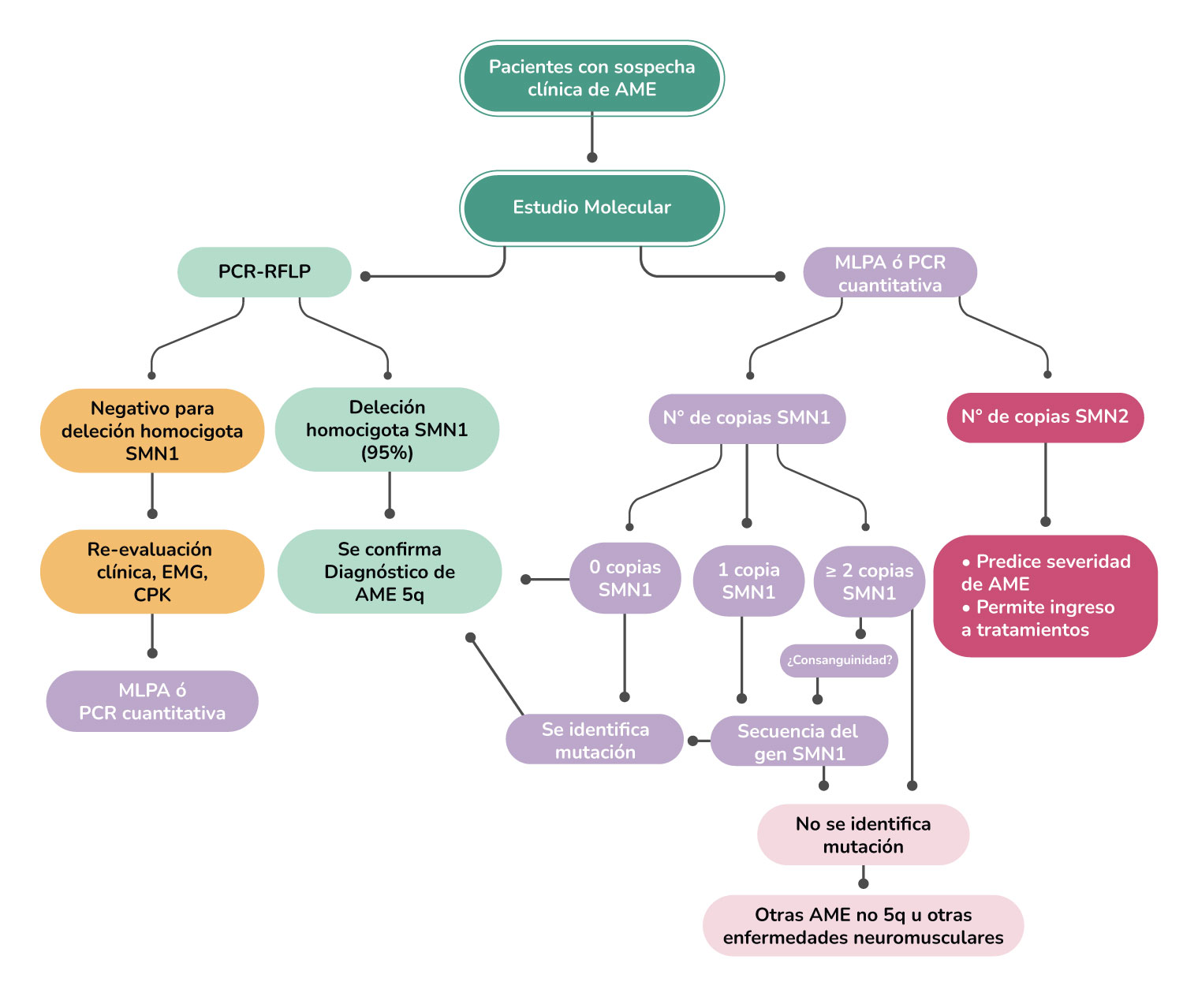
Existen otra serie de pruebas que pueden ayudar a establecer el diagnóstico:
Técnicas basadas en PCR
• La reacción en cadena de la polimerasa, conocida como PCR por sus siglas en inglés Polymerase Chain Reaction, es una técnica de biología molecular cuyo objetivo es obtener un gran número de copias de un fragmento de ADN.
• Una de sus principales aplicaciones es la realización de pruebas genéticas para detectar mutaciones en el ADN que puedan provocar algún tipo de enfermedad.
• Se pueden hacer análisis del ADN de los padres para ver si son portadores, o analizar el ADN de los hijos por si están afectados por una enfermedad hereditaria.
Estudios de velocidad de conducción nerviosa (CMAP)
• Es una prueba que mide la rapidez con la que el impulso eléctrico se desplaza a través de un nervio para ver si está funcionando correctamente. Puede dar información sobre alteraciones tanto de los músculos como de los nervios.
• Otras pruebas incluyen: análisis de sangre (incluye enzimas en suero, sobre todo la creatin- fosfoquinasa o CPK que puede estar elevada en estos pacientes) y análisis de orina.
1. Fundame
2. Ninds.nih.gov
3. Ninds.nih.gov
4. Roche.com
5. Guías de Atención Pediátrica (GAP), Hospital Garrahan
• Belter L et al. Journal of Neuromuscular Diseases 5, 2018;167–176.
• Bowerman et al. Disease Models & Mechanisms, 2017;(10):943-954.
• SMA Europe • Sugarman EA, Nagan N, Zhu H, et al. Pan-ethnic carrier screening and prenatal diagnosis forspinal muscular atrophy: clinical laboratory analysis of > 72,400 specimens. Eur J Hum Genet. 2012;20(1):27-32.
• Prior TW, Finanger. Spinal Muscular Atrophy. GeneReviews. December 22, 2016.
• Rarediseases.info.nih.gov
• ncbi.nlm.nih.gov